Bridging the bandgap
How scientists use entropy to power a better future
A vast expanse with plenty of potential
To effectively make the transition away from fossil fuels and toward renewable energy, we need batteries that charge faster and store more energy. We need more efficient solar cells and cheaper computer chips, too. We need TV screens that waste less energy, plastics you can more easily recycle, and catalysts to speed up important chemical reactions. Improvements in all these areas are urgent, and one common discipline is central to them all: materials design.
Scientists rely on theory, experience, and chemical insight to design and synthesize new materials with the properties necessary to make our most critical devices work efficiently and effectively. They scour the scientific literature, noting the syntheses that work as well as those that don’t; they take advantage of their own intuition alongside the calculations of computational models to determine which precursors to use and the methods by which to combine them. Yet, as many of us may have thought at some point during a chemistry laboratory class, this problem might just be too hard for us. The number of possible materials that one could make is mind-bendingly large on its own. If we multiply that number by the possible synthetic routes one could take to reach each of those materials, the number becomes unfathomable. Even the most dedicated graduate student could never honestly state that they have fully optimized a materials synthesis.
The compositional space of materials has always been enormous, but it hasn’t necessarily been a problem. Historically, the development of specific and useful materials did not require searching through every possibility. Generations of materials scientists have identified simple materials with a limited number of components and desirable properties. Then, over decades, they have systematically, gradually, and successfully improved these materials to create the foundation of all our modern technologies. But some scientists suggest that diving into the complexity and the enormity of the compositional space is exactly what is needed to address our most pressing issues in energy storage and production.
The concept of high-entropy materials has been gaining interest for the past 20 years, with a recent focus on how these materials can be utilized for energy applications. Entropy can be thought of as a measure of disorder, but also of complexity. For instance, a high entropy material might be composed of several different components (often five or more) in roughly equal proportions. This would be an example of a high compositional entropy. In a high entropy material, different components could be equally likely to occupy a given position. This would be an example of high configurational entropy. This disorder might not sound appealing when designing a material, but the complexity also provides a materials scientist with more knobs to fine-tune functionality. As an example, an expert explained to me why a lack of precise control is sometimes exactly what is needed to improve a specific and vital function.
Leveraging disorder for a better battery
Yan Zeng was a staff scientist in Gerbrand Ceder’s group at Lawrence Berkeley National Laboratory (LBL), and in January she started a position as assistant professor of chemistry at Florida State University. She walked me through the counterintuitive notion of how intentionally creating disorder can lead to a better material for a battery. A battery essentially works by transferring an atom with a positive charge, an ion, from one piece of metal to another through a material called an electrolyte. When the battery is connected to a circuit, the positive charge moving through the electrolyte allows for a negative charge to move through the wires, thus powering your device. Similarly, to recharge the battery, it needs to be connected to a circuit that forces the ions to move back in the opposite direction. For a battery to charge and discharge quickly, ions need to move quickly through the electrolyte. A good battery should also be able to store a lot of energy in a small amount of space. Using a solid material as an electrolyte can afford this high energy density, but ions may have a difficult time moving through the stiff solid. Therefore, increasing the speed at which ions can move through a solid material, what scientists would call ionic conductivity, is vital.
In a crystalline solid, one where the atoms are organized in space in a regular pattern, small ions will occupy specific sites in between the other atoms. When induced to move across the solid, the ions will hop from site to site, and the rate of hopping—the time an ion typically has to wait before it makes a hop—will determine how quickly the ions move. The rate of hopping generally depends on the typical energies of the ion both in the site from which it starts and in the site to which it is going. For instance, if in one site the ion is very stable and typically has very low energies, the rate of hopping will be very slow toward a site where the ion is unstable and typically has very high energies. However, if there are two adjacent sites with similar energies, the rate of hopping between them will be quite fast.
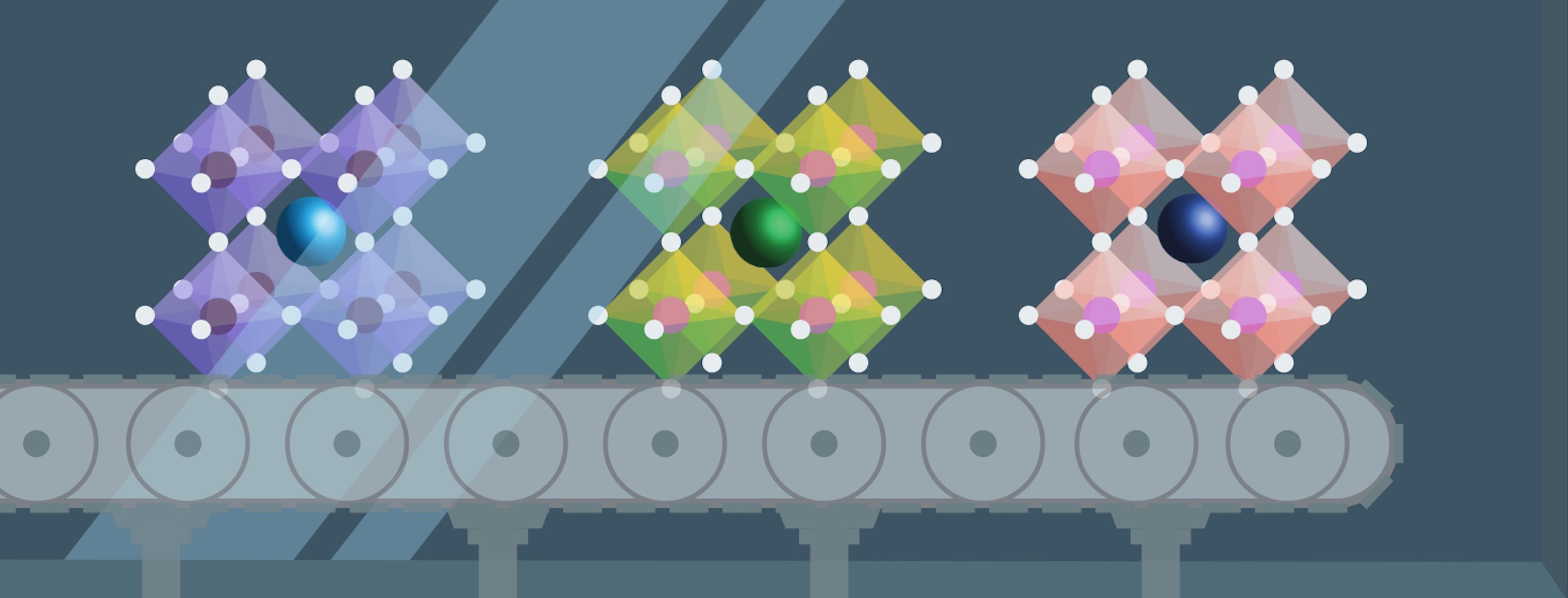
In Professor Zeng’s and her collaborators’ experiments, they created a high entropy crystal where the same site on the crystal was randomly occupied by different metals of different sizes. She describes this “size effect” on the material, explaining that “different [metal atoms] have different radii and combining them will create an uneven lattice distortion. So, some places in the crystal will shrink and other parts may be expanded a lot.” This uneven shrinking and stretching changes the typical energies that an ion has when it occupies sites in different parts of the material. In general, it isn’t obvious that this distortion would lead to fast pathways for ions through the material. But when the energies of all the sites change, it is almost guaranteed to create at least one fast pathway through the material along which the energies of the sites are all similar. Somewhat miraculously, this one pathway is enough to increase the ionic conductivity of the whole material. Professor Zeng explains the phenomenon with an analogy: “So you can understand it as like a highway. Say we want to go from point A to point B and there are multiple streets and roads and one highway. It doesn't matter if there are some streets that are blocked. It only matters that we have one highway where there is no traffic.” Professor Zeng and her collaborators first tested their idea of altering the site energies through distortion in a computational model, then realized the system experimentally, showing in several examples that the ionic conductivity did increase significantly when disorder was introduced to the material.
The size effect that Professor Zeng describes is general and can be easily explained and applied in other materials, but there is another critical factor that affects ionic conductivity: the chemical effect. Professor Zeng explains that the chemical identity of the metal atoms used to make up the lattice will also naturally have an effect on the ionic conductivity that is just as large as the size effect. However, this effect isn’t as easy to understand or predict. While chemists learn trends and materials scientists often have an incredible sense of chemical intuition, predicting how a certain combination of four or five metals will interact to influence the ionic conductivity of a material is a daunting task. Theoretical methods can attempt predictions of material properties, but the guaranteed way to know how a material behaves is to make it. Therefore, to fully explore the compositional space of materials, one focus must be on making the syntheses as easy and efficient as possible.
Turn the heat off, I’m not made of money
While disorder might seem like it would be difficult to avoid when making a complex material, in some contexts it can be a challenge to induce it. We can explain this with a quick trip to general chemistry class (very quick, I promise!). The preferred state for any material is determined by something called its “free energy.” If we start with some precursors and mix them together to form a product, the total free energy change will be a balance between the change in enthalpy (a measure of the strength of the new chemical bonds compared to the old ones) and the change in entropy. The catch is that often the state with the optimal enthalpy and the state with the optimal entropy are very different. If you mix some components together, the state with the optimal enthalpy is likely going to be one where the different components separate into their own domains, while the state with the optimal entropy is the one where all the components are completely mixed together. If even that amount of general chemistry made your eyes glaze, we can think of this competition between enthalpy and entropy like a wedding with lots of groups of people who don’t know each other. We are aiming for the “wedding state,” where everyone is dancing, mingling, and having a great time. But we know that, if we aren’t proactive, each small group will just stand in a circle and talk amongst themselves. So, we have to change our experimental protocol. If we open up the bar, put Single Ladies on the speakers, and get things going with our best approximation of putting a ring on it, we might just be able to coax everyone out of their groups and onto the dance floor.
And it turns out chemists have their versions of a Beyoncé classic as well. By mixing a larger number of components together, you can make the potential entropy change large enough so that it can beat out the enthalpy. Still, even if the high entropy state is preferred, we can still get trapped along the way to your product in states with low entropy but very strong chemical bonds (like when a slow song comes on and everyone begins to migrate back to their tables). The simplest way to combat this problem is by raising the temperature during the synthesis (maybe not at the wedding). At higher temperatures, strong chemical bonds will break more easily, and we will be much more likely to reach our high entropy product in the end. In a sense, a high temperature emphasizes entropy at the expense of enthalpy.
Often the syntheses of traditional high entropy materials are conducted at temperatures on the order of 1000 °C, but this poses a problem for using high entropy materials in devices. If the syntheses require very high temperatures, there is more energy wasted in manufacturing, resulting in higher costs across the board. To manufacture at scale, scientists ideally want a synthesis that doesn’t require extreme conditions and is cheap and simple to perform. Researchers in Professor Peidong Yang’s group at UC Berkeley have recently demonstrated such a high entropy synthesis.
A graduate student in the Yang group, Yuxin Jiang, is working to synthesize high entropy metal halide perovskites without resorting to high temperatures. Perovskites are popular materials mainly because of the unique way that they interact with light. They have the potential to make devices such as solar cells and light emitting diodes (LEDs) more efficient, so it's important to be able to tune this interaction with light. One way to do this is by changing the metal in the metal halide perovskite. In the high entropy material synthesis, Dr. Maria Folgueras, Jiang, and their coworkers were able to make metal halide perovskite crystals in which the metal site was occupied by six different metal atoms in nearly equal ratios. Importantly, their synthesis can be done at room temperature and is a very simple protocol in which powder precursors are mixed in a solution and the crystallization occurs during the slow evaporation of the solvent. Jiang explains that part of the reason this easy synthesis was possible was because “halide perovskites, compared to a lot of other materials, have an advantage of a relatively lower cohesive energy, which is the energy that you need to separate different types of atoms in order to put them back together.” Because this cohesive energy is lower, you do not need to raise the temperature very high for the material to be able to “find” its minimum free energy state.
The ease and simplicity of this type of solution-based synthesis opens the door for a more efficient exploration of the properties of this class of materials. Jiang hypothesizes that this method “could create hundreds or even thousands of single crystals of different elemental ratios.” But there is a question that naturally arises: how do you characterize and categorize potentially thousands of products that all might have different properties? The Yang group and many other researchers continue to study the optical properties of self-assembled halide perovskite crystals and to improve the synthetic methods. Another member of the Yang group, Cheng Zhu, described how they are building on “40 or 50 years of previous studies to combine old knowledge with the new material systems to get all kinds of different structures and assemblies.” But, 40- or 50-years’ worth of papers might be too many for anyone to read, so how can we really leverage all the knowledge that’s available to us?
AI to the rescue (hopefully)
You might need to read every scientific article on the internet to feel confident in predicting the function of a complex material that’s never been synthesized, but luckily most of us are already familiar with programs that soak in every piece of information they can find on the web. Large language models seem ubiquitous to us now, and it turns out one can use them just as successfully for science as they can for writing emails and tricking people on social media. Researchers in the Ceder group, as well as Professor Kristin Persson’s group at LBL, have developed software that scours the scientific literature and uses the data it finds to suggest possible recipes to obtain materials that have never been made before. The software uses patterns it finds from syntheses described in the literature as well as data on the stability of various compounds obtained through computer simulations to be able to select appropriate starting materials and map out all the necessary steps to reach a target material. A postdoctoral researcher on the project, Matthew McDermott, described the algorithm as “figuring out the right angle to take in the very high-dimensional space of all the possible phases that exist.” The team at LBL has now integrated this software with a fully automated lab so that, when a recipe is proposed, robots can follow it and (ideally) make the material without any human intervention. The first rigorous tests of this workflow were documented in Nature in what postdoctoral researcher Nathan Szymanksi referred to as “version zero of the A-lab,” where the “A” stands for autonomous. In this version, the researchers focused on the synthesis of crystalline materials via a simple procedure in which powder precursors are mixed together and then heated in a furnace to react. They attempted to synthesize 58 materials, 52 of which had never been synthesized before to their knowledge, and none of which were used in training the machine learning program. The assessment and characterization of the products was also completely automated using X-ray diffraction, a foundational technique in materials science which provides structural information on a material. There is yet another computational model to interpret the results of the measurements, so that no human needs to be involved. If the synthesis results in less than 50 percent of the intended target, the model uses the information from the failure to propose a new recipe and the robots try again. They repeat this process until the synthesis is a success or until it fails five times in a row (at that point, the researchers consider it a waste of resources to keep trying). Ultimately, they were able to make 41 out of the 58 targets by following this procedure.
Researchers were careful to emphasize that the project is still in its initial stages and has significant room for improvement, but they eagerly anticipate the future impact of the autonomous lab. Dr. Szymanski described one of the failures in synthesis that actually led to an important correction on the computational side of the project. A particular compound was predicted to be highly stable by the model, but the A-lab was unable to make it upon multiple attempts. This caused researchers to investigate further, and they discovered an error in the computational model that led to an incorrect prediction of the energy of the compound, as well as many other similar materials. The researchers noted that catching this error likely made the materials database they were using significantly more accurate. Dr. McDermott also pointed out that often the starting materials for a synthesis are chosen for convenience when humans do the selection. He thinks “we can do a lot better with the precursors we use if we're willing to step outside of what is convention,” and he believes the A-lab has the potential to do this. The computational model has some limitations since it has been trained on syntheses that have already been attempted by humans, but because it can also predict the stability of phases that haven’t already been synthesized, it has the capability to propose novel synthetic routes.
What to do with a robot-filled future
The initial tests of the A-lab focused just on synthesizing predetermined targets, but, looking toward future efforts, Dr. Szymanski explains, “You could extend a lot of [the A-lab] to do further characterization, measuring device performance to figure out whether it is a good battery or some other technology, and that would really automate the whole process end to end.” Professor Zeng noted that the current A-lab project is not currently connected to the high-entropy battery projects mentioned earlier in this article, but that connecting the two could certainly be a goal for future autonomous labs.
All the researchers are confident that autonomous labs will become much more common in the coming years. Dr. McDermott mentioned that, at our current level of technology, some tasks in the laboratory might still be more efficiently performed by humans, but the advantages of automation for syntheses like those performed in the A-lab are clear enough that he has “no doubt that most chemistry groups will incorporate automation in some way.” Professor Zeng also touched on the prospect of machine learning making the syntheses themselves more efficient. She says she believes “there can be a model that not only considers if it’s feasible to make the target but also considers other factors like the costs of energy consumption or the cost of precursors or the cost of time.” In the final moments of our conversation, Professor Zeng also emphasized the prospect of using the autonomous lab as a way of increasing scientific transparency and sharing data more effectively. She highlighted three types of data that ought to be shared for the success of autonomous laboratories and the design of complex functional materials. One, she says, “is the results of what has been produced, including both successes and failures.” The second is “the machine learning algorithms or training sets so that other groups might make further improvements and adjustments to the model.” And the third is “the knowledge of how to build this kind of lab and how to implement this AI and robotic driven workflow.” Surely sharing the knowledge we have gained is the best way to leverage the new and quickly evolving technological resources of the scientific community. Using these tools to lean into complexity and leveraging disorder has promise for materials design, and we can only hope that it helps us to address our urgent climate emergencies.