A paper crane appears impressively intricate, especially to a novice origami maker struggling with the first creases. But fumbling hands and crumpled paper belie a different type of folding expertise. Imperceptibly, legions of molecules inside the origami maker’s body constantly confront a much more complex folding task. These molecules, called proteins, reliably fold into one out of an enormous number of possible structures in a fraction of the time it takes to make a paper crane. With no hands to guide it, each protein molecule must traverse the pathway to its correct shape with superhuman speed and precision. And while poor origami technique results in wasted paper at worst, the consequence for failed protein folding can be death.
The mystery takes shape
A protein is a biological machine composed of smaller molecules called amino acids that are linked together in a chain. The backbone of this chain is made up of the main chain atoms of the amino acids, while the other atoms in each amino acid, called the side chain atoms, protrude from the main chain like charms on a charm bracelet. It’s the side chains that give each of the 20 or so amino acids its unique properties, and the sequence of amino acids defines each protein’s specific structure and function.
The way a particular amino acid sequence encodes the structure and function of a protein isn’t entirely understood. And while it takes several minutes to learn to accurately replicate the paper crane, understanding the path a protein takes to achieve its folded structure has vexed researchers for decades. As early as the 1960s, when researchers first developed techniques to determine protein structures, they realized that understanding protein folding would be far more difficult than they had initially anticipated. Humans have thousands of different types of proteins, and each one, researchers found, has a unique structure. More perplexing still, these proteins fold into a multitude of shapes that researchers had not predicted.
When researchers could not come up with satisfying explanations for the origins of proteins’ complex shapes, excitement and controversy followed. They knew that a newly manufactured protein molecule has all the structure of an overcooked spaghetti noodle. But, defying intuition, the mostly unstructured new protein wriggles around like a noodle in boiling water, contorting itself into an extremely specific, elaborately organized three-dimensional structure called the native state.
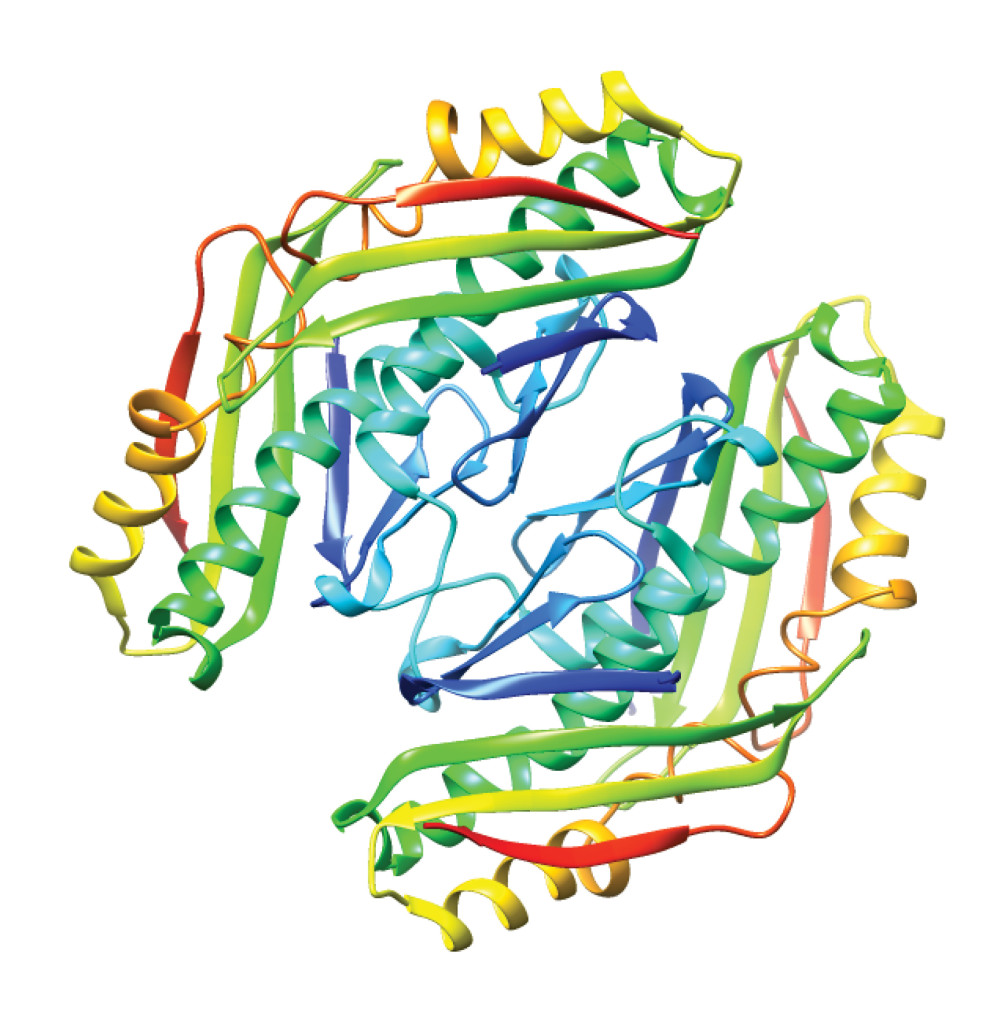 Expansive structural diversity influences protein function. PDB ID: 1vla.
Expansive structural diversity influences protein function. PDB ID: 1vla.
Finding the native state is critical since it allows each protein to perform its specific biological function. For some proteins, failure to reach the native state may cause disease or cell death.
In rare cases, protein misfolding can be fatal for an entire organism. Even a subtle nudge down the wrong folding pathway can lead to the formation of dangerous protein aggregates implicated in maladies such as Alzheimer’s and prion diseases. The fact that any of us are alive is a testament to the fact that proteins typically manage to get the folding process right. Yet after decades of intense scientific inquiry, researchers have just begun to unravel the mysteries of protein folding, and it seems that nature has tricks at every turn.
The missing link
Only under the guidance of a sentient artist can a paper crane take shape, but the rules are different for origami on the molecular scale. A protein molecule folds into its native structure unsupervised by humans—but not necessarily without help. Many proteins fold with assistance from cellular components called chaperones, which are present in all types of organisms. Chaperones come in several varieties—some use energy to speed up folding, while others provide a safe capsule where individual protein molecules can sample different folded shapes, minimizing the chance that misfolded or partially-folded proteins will be flagged for destruction or clump together to form harmful aggregates.
But the existence of chaperones doesn’t solve the protein-folding problem. Most chaperones do not influence the final structure a protein assumes, and not all chaperones speed up folding. Recent research also hints that even when chaperones should be most vital for folding, they may not actually be essential. A cell normally responds to a high-temperature environment by increasing its number of chaperones, which can help prevent the heat from disrupting proteins’ structures and causing them to aggregate haphazardly. However, Andrew Dillin’s lab in the Department of Molecular and Cell Biology at UC Berkeley recently showed that preventing some cells from making more chaperones during heat shock isn’t lethal unless other critical cell functions are also blocked.
Complicating matters further, chaperones are also proteins, so invoking their existence to explain all protein folding would generate a chicken-and-egg conundrum. Although some proteins require chaperones to fold, others can carry out the entire folding process without assistance from any cellular components. When researchers force these autonomous folders to unfold, the proteins are often able to completely recover their shapes and functions.
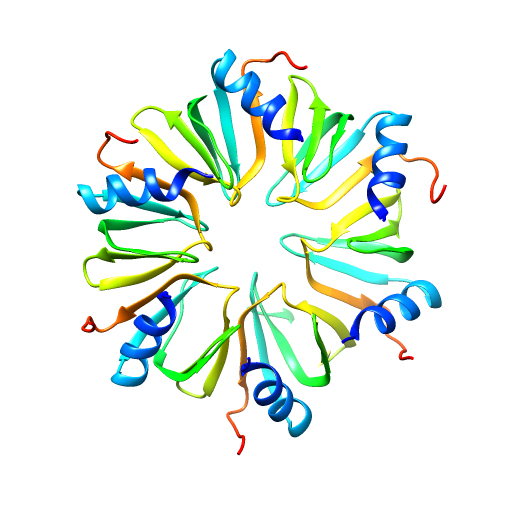 Expansive structural diversity influences protein function. PDB ID: 3qhs.
Expansive structural diversity influences protein function. PDB ID: 3qhs.
Autonomous folders don’t just refold perfectly—they refold quickly. The entire refolding process is usually complete within seconds, and the fastest-folding proteins regain their active forms in just millionths of a second. With their uncanny ability to rapidly solve folding puzzles that stump human scientists, the autonomous folders are the muses of protein folding researchers. To learn how some proteins so swiftly solve the folding problem, scientists need to understand more than just proteins’ structures: they need to dissect the autonomous folders’ techniques.
In almost all cases, the stability of a protein’s native state surpasses that of the unfolded state and the various possible misfolded structures. But the stability of the native state doesn’t explain how proteins fold so quickly. Even favorable reactions can proceed so sluggishly that they are almost unobservable. Graphite, for instance, is a more stable form of carbon than is diamond—diamonds don’t actually last forever. Instead, diamonds decay into graphite, although the process takes millions of years because there is no fast route for the carbon atoms to rearrange from one form to the other. For proteins, too, the transition from the initial structure to the most stable one requires a massive rearrangement. This implies that a protein should find its native state eventually, but “eventually” is not fast enough in biology.
A paradox on the cosmic scale
Given information about the shape of a finished paper crane but no instructions, an inexperienced origami maker constructs better and better approximations of the crane with each iteration, creasing and uncreasing, slowly sampling various methods. The result may be frustration, but the hobbyist still has the luxury of a sluggish, wandering journey to the end product. Proteins, in contrast, often don’t have much time to reach the native state. Living creatures need proteins to rapidly achieve their functional, folded structures, sometimes in small fractions of a second. Any holdup in the folding process could mean delayed response to stimuli, inability to orchestrate necessary cellular events, or aggregation of less stable, partially folded intermediates, any of which could have dangerous consequences.
Just as origami paper can be folded in many ways, every one of the chemical bonds connecting the thousands of atoms that make up each protein can rotate. In 1969, not long after the beginning of protein folding research, scientist Cyrus Levinthal at the Massachusetts Institute of Technology estimated that each protein could adopt a staggering 10^143 geometrical arrangements. Nearly all of these possible arrangements would bear absolutely no resemblance to the native state, and most would be nonfunctional.
Chemical bonds can rotate much faster than the most expert origami maker’s hands can manipulate paper, but even if proteins could sample trillions of shapes per second, an average-sized human protein would take longer than the current age of the universe to fold. The difference between this predicted rate and the observed folding rate was dubbed Levinthal’s paradox.
Scientists savor the prospect of dismantling a paradox. Protein folding researchers, including Levinthal, recognized that there must be another mechanism to explain how proteins fold so rapidly. One appealing hypothesis is that proteins fold in smaller segments that lock into place after achieving the correct structure. Rather than sampling every shape, the protein would be divided into smaller, autonomously folding sections. According to this model, instead of crumpling up a slightly defective paper crane and starting from scratch, the origami maker would correct the problem by revising the incorrectly folded part while leaving the rest of the structure intact.
Breaking up the task of folding makes sense, and some mathematical models suggest that dividing proteins into smaller chunks would yield reasonable folding rates. Folding proteins piece by piece may resolve Levinthal’s paradox, but this explanation leaves many questions unanswered. In the case of the paper crane, we know which part is the wing and can adjust it accordingly, but proteins lack this simple means of defining which segment is which. Yet in this regard, the protein’s complexity is an asset.
Molecular choreography
An unfolded piece of origami paper is flat and featureless, providing no hints about where to begin and which folds to try. In contrast, biology rigs the protein folding process by building clues about the correct path right into the unfolded protein. The chemical natures of the amino acids bestow each region of the protein with different folding propensities. Some sequences of amino acids tend to form spirals called alpha helices, while others fold into tight loops and turns or line up to make sheets. Information about unfolding rates can inform researchers about folding, and by developing techniques that unfold proteins extremely rapidly, researchers have learned that some parts of proteins’ structures, particularly the alpha helices, fold at a faster-than-average rate. Individual components such as sheets and helices are the building blocks of protein structure, and their spatial orientations give each protein its shape. If some parts of the structure are quick to take form, it could influence the protein’s native state.
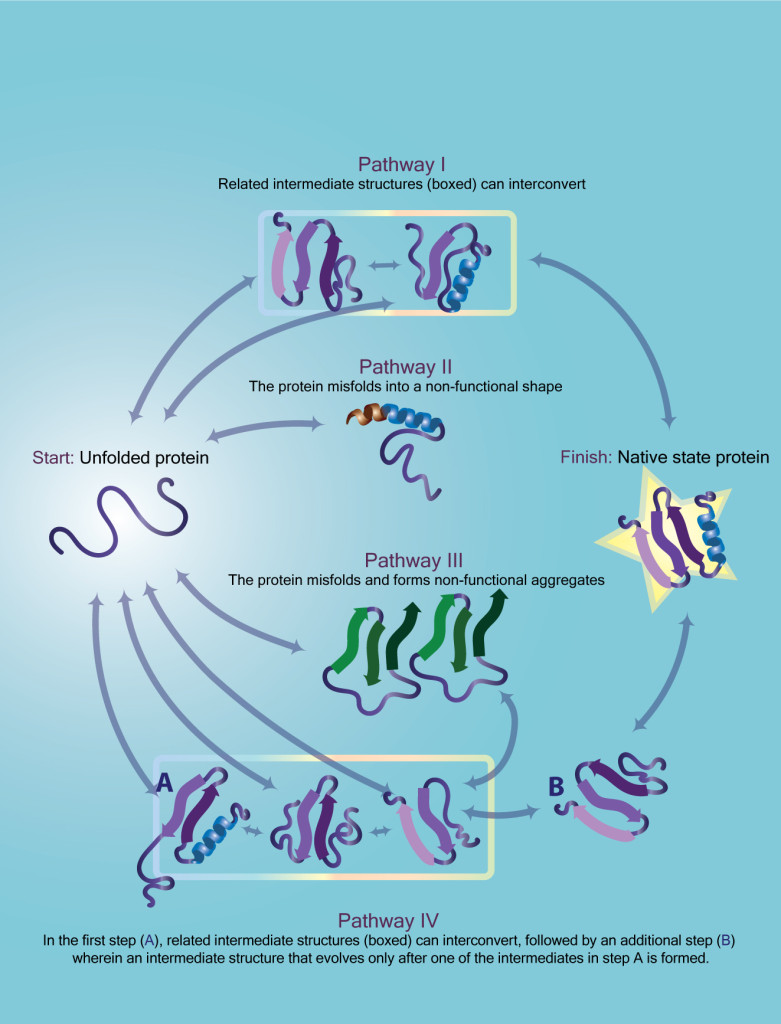 The unfolded protein may predominantly follow one pathway, but it may access other pathways by chance or due to changes in its environment. Some of the alternative pathways may also lead to the native state, here indicated with a gold star, while others could be unproductive or even dangerous. In some cases, intermediates along different unfolding pathways may also be able to interconvert within groups (outlined).
The unfolded protein may predominantly follow one pathway, but it may access other pathways by chance or due to changes in its environment. Some of the alternative pathways may also lead to the native state, here indicated with a gold star, while others could be unproductive or even dangerous. In some cases, intermediates along different unfolding pathways may also be able to interconvert within groups (outlined).
This and other observations make sense in light of the sequential model of protein folding, which states that a protein achieves the correct structure in segments, writhing in a predictable fashion toward the native state. According to this model, each type of protein has a unique path to its specific native state, and every copy of a given type of protein follows the same folding path. Although other models had been proposed, the sequential model made intuitive sense—and seemed to explain much of the experimental data.
When multiple models are capable of explaining experimental data, scientists usually work under the hypothesis that the simplest one is the most probable. Thus, experimenters aim to test the least complicated model for exceptions and contradictions. Sequential protein folding is appealing because it explains protein folding in a simple way, and mathematical models based on sequential folding at first seemed capable of matching most of the experimental data. But there were many who thought the model would need to be revised, expanded upon, or even replaced because they doubted that a protein would always fold via the same pathway.
Susan Marqusee, head of a protein folding research group in the Department of Molecular and Cell Biology at UC Berkeley, says that whether a given type of protein might be able to reach its native state by multiple paths has been a contentious subject for quite some time. “There’s been a longstanding question in the field,” she says, “about whether, when proteins fold or unfold, there’s a single trajectory or a single transition state that they go through. Theoretical studies have always shown that there should be many options, but experiments never reveal that.”
Illuminating the invisible
For all the insight they have provided, the earliest protein folding experiments left much to the imagination, and scientists’ imaginations brew controversy. Many questions about protein folding could have been resolved long ago if not for one glaring blind spot: individual protein molecules are invisible. Each protein molecule is a few billionths of a meter across, much too small to be seen in the act of folding. Undaunted by these obstacles, several teams of researchers have set out to visualize the invisible.
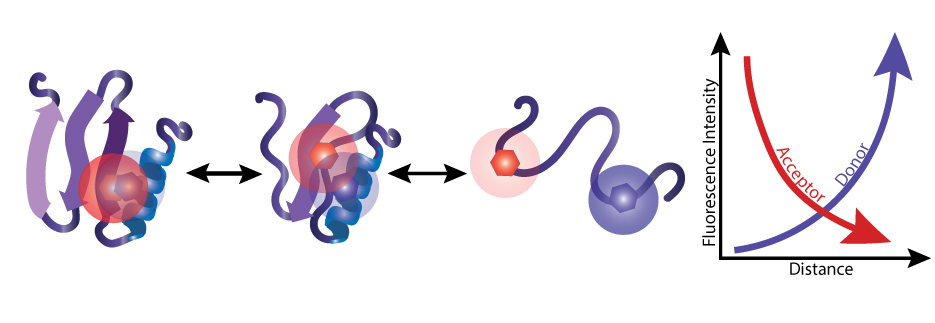 Two specific fluorescent tags, known as a FRET pair, are attached to different sites on each protein molecule. When researchers excite the donor tag (blue), it emits energy in the form of light (fluorescence intensity). If the donor is near the acceptor (red), the donor transfers some energy to the acceptor instead, causing the acceptor to emit its own light.
Two specific fluorescent tags, known as a FRET pair, are attached to different sites on each protein molecule. When researchers excite the donor tag (blue), it emits energy in the form of light (fluorescence intensity). If the donor is near the acceptor (red), the donor transfers some energy to the acceptor instead, causing the acceptor to emit its own light.
In 1997, María Pilar Lillo in the Departamento de Biofísica at the Instituto de Química-Física in Spain worked with collaborators to piece together a movie of the process by pinpointing the location of different parts of the protein caught in the act of folding or unfolding. To accomplish this, the researchers applied a phenomenon called Förster resonance energy transfer (FRET), which is a special type of measurable interaction that depends strongly on distance. Two chemicals that interact in this way, known as a FRET pair, can thus serve as a molecular ruler. The researchers took advantage of this interaction by chemically linking FRET pairs to each protein molecule at sites that are neighbors in the native state but farther apart in the unfolded state. Then, by unfolding the protein while monitoring the change in FRET interactions over time, the researchers could determine which parts of the protein are first to move and what path they take.
These FRET experiments painted a vivid picture of a single protein’s unfolding mechanism, and the same technique was later used to visualize the folding and unfolding of many additional proteins. Lillo’s results suggested that their protein unfolded via a sequential trajectory, stopping at specific, partially unfolded intermediates on the way to a nearly completely unstructured, unfolded state. Even today, protein folding researchers are still gaining new insights from increasingly sophisticated FRET experiments. But just as this technique was first reported, other research groups were developing another method for visualizing protein folding—and what they found would call dogma into question.
Folding proteins bit by bit
By the late 1990s, a revolution in computing had extended the reach of biological simulations. Contributing to the advances were both massive supercomputers and the crowdsourced project Folding@home, which exploits the unused computing resources of volunteers around the world to power its simulations. With these tools at their disposal, computational researchers burst into the protein folding field with simulations known as in silico protein folding experiments. By employing sophisticated modeling of molecular-scale forces, computational researchers could simulate the folding of small, rapidly folding proteins.
Although in silico experiments have many inherent limitations, computational approaches have paired folding rules with human intuition in order to improve predictions of proteins’ native structures. At the University of Washington, researchers in Zoran Popović’s group joined forces with members of David Baker’s lab to create FoldIt, a publicly available puzzle game. In FoldIt, human users use intuition and spatial reasoning to help computers determine a protein’s proper fold based on common structural patterns. The strategy works shockingly well: in a 2009 competition to computationally determine protein structures, FoldIt players made the most accurate model and took first prize. Later, FoldIt players even made headlines by helping to determine a protein structure that researchers had failed to solve for years using traditional methods.
But even before FoldIt and its kin were developed, computational researchers had already begun to uncover information that deepened a growing rift in the field. Results from the earliest simulations to today’s in silico folding investigations depict the way each protein, initially formless, wriggles into a well-defined structure. Beyond their visual appeal, the computational results seemed at odds with the notion that a protein almost always takes the same path to the folded state, one of the key assumptions of the basic sequential model. In an apparent contradiction, repeated folding simulations on a single protein often revealed that several sequences of events could lead to the final, folded structure.
According to simulations, proteins do fold in stages, but each step along the pathway represents not a single state but a whole ensemble of partially folded states. Only a few of the partially folded proteins at each step might be capable of leading to the native state, but rapid interconversion among the various states in the partially folded ensemble would allow the folding protein to stumble toward the correct path. Just as the novice origami maker notices a mistake after a series of incorrect creases and backtracks accordingly, a protein folding along an incorrect path reaches an insurmountable barrier and undoes a step or two, giving it another chance to reach the native state.
All roads lead to Rome
The computational results alone weren’t enough to convince everyone that proteins could take more than one pathway to the native state. A simulation is only as accurate as the human-supplied parameters used to guide it, Marqusee explains. “That’s the limitation right now for both of us [experimental and computational researchers]—there isn’t a direct tie between the two,” Marqusee says. Computational researchers can provide ideas to be tested by experimenters, who then generate more data that can be used to formulate better simulations.
Although the findings from simulations don’t stand alone, they spurred more serious experimental investigation of the possibility that proteins might have many ways to reach their native states. Even before the first folding simulations, there were isolated experimental reports that described proteins folding via multiple pathways, Marqusee says, but “they were considered oddballs.” Indeed, some of the most enticing experimental evidence that proteins could traverse multiple folding pathways had been seen in a protein exposed to a high concentration of a denaturant, a type of chemical that unfolds proteins. These harsh conditions are unlike those found in live organisms where real biological phenomena—and, critically, protein folding diseases—occur.
While some had reservations about the general applicability of experiments that suggested multiple pathways, others went as far as to question how relevant discoveries of multiple pathways might be. “There are always people who say, of course there could be multiple ways to fold,” Susan explains. “But if the protein predominantly takes this one way, what does it matter?” Intuitively, it makes sense that a protein might have more than one way to reach the same native state. One method of making paper cranes might involve folding the wings first, while another begins with the tail and body. As long as every segment assumes its proper position eventually, is the number of possible ways to reach the same outcome really important?
One molecule at a time
For researchers to find evidence that proteins can take many paths to the native state—and that such a discovery would be biologically relevant—they needed to take an approach completely different from that of conventional protein folding experiments. Most of the existing experimental methods used to study folding require significant perturbation of the protein. This problem caught the attention of researchers Emily Guinn and Bharat Jagannathan in Susan Marqusee’s lab, and they had a new approach in mind.
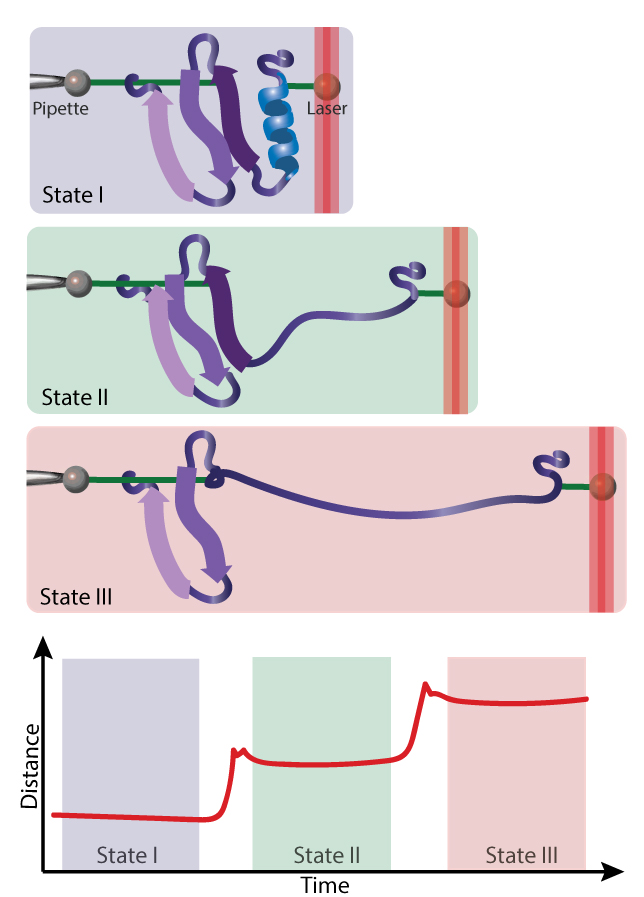 A protein molecule is tethered to two beads. One bead is held in place by a glass micropipette, and the other is pulled by a laser. When this force is strong enough, part or all of the protein unfolds.
A protein molecule is tethered to two beads. One bead is held in place by a glass micropipette, and the other is pulled by a laser. When this force is strong enough, part or all of the protein unfolds.
Guinn and Jagannathan unfolded single protein molecules by mechanical force using optical tweezers, a tool repurposed to study folding by prior Marqusee Lab members in collaboration with researchers in Carlos Bustamante’s lab, also in the Department of Molecular and Cell Biology at UC Berkeley. The researchers captured individual proteins by attaching two sides of each molecule to glass beads caught in a laser beam’s path. The laser traps the beads, stretching the protein like a rope in a game of tug-of-war, so the experimenter can increase the force on the protein by adjusting one laser to drag the beads further apart. At a sufficient force, the protein stretches too far to maintain its structure and unravels.
The Marqusee Lab researchers used the optical tweezers to gently tug on a protein with a minuscule stretching force of just 10 to 30 piconewtons, or about 100 billionth of the force of gravity on an apple here on Earth. “We were using a much lower denaturant concentration and relatively low force,” lead author Emily Guinn says. These subtler disruptions act more like the normal challenges that proteins face in the cell. According to Guinn, “Physiological forces on proteins are very small, on the piconewton scale, so we were looking at forces that are close to that.”
Others have studied protein folding using similarly small forces, but the Marqusee Lab researchers were the first to combine force unfolding and chemical unfolding data for a single type of protein. The combination was critical because each of the methods provides different information about the unfolding transition. Using their combined approach, they discovered that the unfolding pathway when the protein was stretched in one direction was likely the same as the unfolding pathway caused by denaturants.
If the researchers had stopped there, they might have concluded that unfolding always follows the same series of steps, at least for this protein. But when they stretched the protein in another direction, Guinn says, “our data indicated that the protein accesses two new pathways that hadn’t been seen before in experiments.” Specifically, the protein took one pathway at lower forces but took a different route when the force was increased. This provides very convincing evidence that new models of protein folding must incorporate the possibility of multiple pathways. Beyond the theoretical impact, the fact that such a minor environmental disturbance can significantly influence which folding pathway a protein is likely to take has monumental implications.
“What we see is that, in some cases, just a small change in the conditions can alter the folding trajectory,” Marqusee says. “So that means that you might have a different folding trajectory in different cells or even in different regions of the same cell.” Far from being homogeneous, the interior of the cell is composed of diverse microenvironments, varies by cell type and developmental stage, and changes in response to stimuli. If the barriers separating different pathways are small, as the Marqusee Lab’s results predict, any of these factors may influence the mechanism of protein folding in a given cell at a given time.
The future of folding
The potential implications of this new insight for protein-folding diseases are especially tantalizing. For instance, we know that genetic differences cause some people to have variants of normal brain proteins that predispose them to developing Alzheimer’s disease. However, not everyone with these variants manifests the illness. If small differences in the cellular environment alter the pathways by which these variant proteins fold, the result could be that the proteins are more likely to be misfolded or trapped in unstable, aggregation-prone intermediates. If we could understand these other potential risk factors, they might provide clues about how to predict, prevent, or even cure protein-folding diseases.
While curing protein-folding diseases is still a distant goal, improving our current understanding of protein folding is already paying dividends. With a detailed understanding of the construction of a paper crane, the intrepid and artistic might set out to create modified paper cranes, paper frogs or paper planes. Likewise, with an understanding of the principles of protein folding, scientists have been able to modify existing proteins to give them new, desirable functions. Some have even designed proteins completely from scratch.
Others have extended the protein-folding paradigm to nonbiological polymers called foldamers, which have already shown promise for uses ranging from antimicrobial agents to artificial lung surfactants that help premature babies breathe. With these applications already within reach and many others on the horizon, it is clear that the study of the journey of these invisible, squirming biomolecules has the potential to yield great benefits for humankind.
As we have gained understanding of how proteins fold, the scope of protein-folding research has expanded to encompass proteins’ behavior, movements, and structural fluctuations. “That to me is protein folding,” Marqusee says. “It’s the barriers between each possible state and everything else in between.” When scientists solved the first protein structures—mere snapshots of a protein’s dynamic existence—no one could have known how complex the protein-folding problem would become. But if the findings so far are any indication, there may be even more to this molecular origami than we can ever understand.
This article is part of the Fall 2015 issue.