The human drive to categorize things is an impulse that’s hard to suppress. In one famous example, Plato declared that man was "an animal, bipedal and featherless," receiving great public acclaim. Diogenes the cynic cleverly rebutted this claim by displaying a plucked chicken and proclaiming, "Behold! Here is Plato’s man." Afterward, or so the story goes, Plato appended the definition to include "with flat broad nails."
Though comical, the allegory illustrates that taxonomy - the practice of finding, describing, and classifying organisms - is certainly an iterative sport. For most of human history, these classifications were based on morphological traits visible by eye or microscope. However, pitfalls abound when morphology is the sole criterion. After all, dogs belong to a single species but they certainly aren’t all cast from the same mold. By contrast, the deep sea floor is teeming with genetically diverse species of bacteria that all look pretty much the same.
These days, even the term "taxonomy" is a relic of the past, conjuring up visions of stuffed birds stashed in the dusty archives of natural history museums. The modern scientific practice - often known as phylogenetics - has evolved from taxonomy per se by its heavy reliance on using evolutionary relationships between species as the basis for classification. The end product of these investigations is often an evolutionary tree, a branching diagram showing the relationships between different species or genes. In fact, some of the first trees were drawn by Charles Darwin and were popularized by The Origin of Species.
Inferring evolutionary relationships is no easy task, especially in vast swaths of the tree of life that have no fossil record. That’s where DNA can contribute; this latest iteration of taxonomic refinement relies heavily on sequencing genomes. In the past few decades, increasingly sophisticated molecular technologies have yielded a bounty of genetic information that speaks volumes about the relationships between species and their evolutionary heritage. Though the DNA transcript is traditionally perceived simply as the blueprint for proteins, it turns out that a compelling but tangled historical narrative is also scribbled into its margins.
The root of the story
Many key chapters have already been deciphered. One of the first examples hearkens from the dawn of genetic techniques in the 1970s. At the time, Carl Woese and his lab members at the University of Illinois set out to map evolutionary relationships between bacteria. They focused their attention on the sequence of ribosomal RNA, an essential component of the protein-assembling machinery in all cells. Because rRNA is so essential for life, its sequence changes very slowly, thus allowing comparisons between organisms that span billions of years of evolutionary time. In those days, the technology for sequencing nucleic acids was rudimentary, requiring tedious work to reconstruct the long 1500-nucleotide sequence from shorter sequences of six to 20 nucleotides. Woese was one of only a handful of people who could read the films necessary for deciphering the sequences. After a year of such labor, Woese stumbled upon a radical evolutionary schism in the world of bacteria. His results produced two very divergent sets of rRNA sequences, so different that he was compelled to partition bacterial life into two separate domains: Eubacteria and Archaea. Though recognition of his contribution was slow, Woese’s initially controversial view is now universally accepted.
Applying similar modes of reasoning and vastly improved genetic tools, researchers at UC Berkeley have begun to probe other important transitions in our evolutionary history. These scientists strive to answer important questions about how multicellularity arose in animals and how eukaryotic life arose from the prokaryotic pool by sequencing entire genomes of organisms that have diverged around the time of these transitions. While shedding light on some questions, they have also uncovered many more.
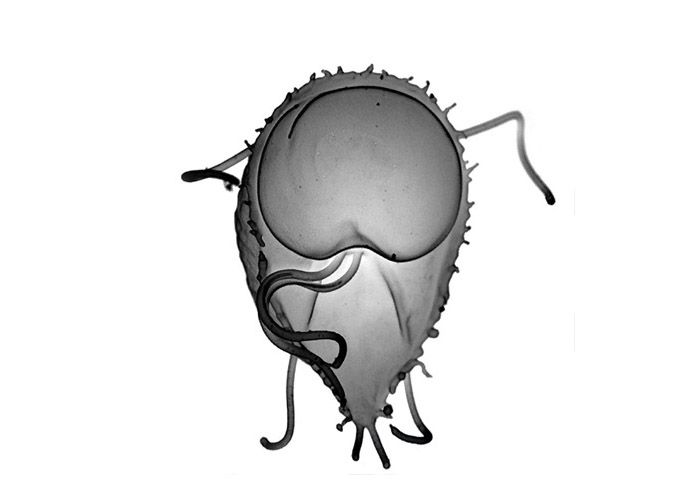
 Naegleria in its flagellate form. The cell's DNA is shown
Naegleria in its flagellate form. The cell's DNA is shown
The independent lifestyle
That’s where the sequence of Naegleria can contribute. Naegleria is considered a highly divergent eukaryote, meaning it’s so different from other types of eukaryotes that the lineage must have branched off very early in eukaryotic history. The lab strain was originally isolated from the eucalyptus grove abutting the Life Sciences Addition, but the species is ubiquitous across soil and freshwater habitats all over the world. Unlike its brain-eating cousin N. fowleri, which can cause fatal meningoencephalitis for unfortunate lake swimmers, N. gruberi is harmless. Its claim to fame is its locomotive versatility; it can reversibly switch from an amoeboid form to a flagellar form in less than an hour. Most importantly, as Naegleria is free-living and not a parasite, it is the first early eukaryote to be sequenced that did not have the luxury of shedding unnecessary portions of its genome.
Naegleria‘s independent lifestyle enables it to contribute significantly to our understanding of the central repertoire of eukaryotic genes. Using data from Giardia, only about 500 gene families were thought to be eukaryote-specific. Careful analysis of the Naegleria sequence increased this number to over 4,000. Over 40 percent of these genes have no recognizable homologs in prokaryotic ancestors, confirming that they must be novel inventions.
Its genome sheds light on the unifying features of eukaryotes, but is still relatively silent on what the evolutionary route to prokaryotes actually looked like. At the end of the day, Naegleria is still very much a true eukaryote, with all its hallmark features. A true intermediate between prokaryotes and eukaryotes, such as a eukaryote with a bacterial cell wall (plant cell walls are different) or a naked Eubacteria without one, would be ideal for solving this puzzle.
Shaping the topiary
Such a chimera has not been yet been found. One reason is that the transition may not have actually occurred, at least not with the linearity we suppose. "The prokaryotic-to-eukaryotic transition has become kind of a dirty word," says Lillian Fritz-Laylin, the graduate student spearheading the Naegleria research.
First of all, true transitional forms may not exist because all eukaryotes have been evolving for the same period of time since the last common ancestor. Thus, Naegleria has been evolving for just as long as humans have been. We may like to think that it, or some other modern organism, has stayed true to the ancestral eukaryote, but this is unlikely. "Whatever happened, happened so long ago, you can’t actually find any transitional forms," says Cande.
Eukaryotes have features stemming from both Eubacterial and Archaeal roots, inspiring two alternative theories about how eukaryotes arose. Some advocate the theory of the blessed event, in which some Eubacteria and Archaea fused to form eukaryotes, contributing the cytoplasm and nucleus, respectively. The alternative theory is that because eukaryotes, Eubacteria, and Archaea are equally divergent from each other, the predecessors to all three groups split off at around the same time. In fact, this kind of trifurcation might even be an oversimplification. Unicellular organisms often reproduce asexually, opting instead to pick up genetic material from the environment to increase genetic diversity, a practice known as lateral gene transfer. For these organisms, the definition of a species is loose. The widespread occurrence of lateral gene transfer means that some evolutionary "trees" can look less like well-ordered branches and more like a tangle of yarn.
Because this transition or trifurcation happened over a billion years ago, any evidence is hazy. Genome sequences give us a better and more complete picture, but they don’t change the fact that the evolution of eukaryotic life may have been a messy, complicated affair, with few known modern descendants of the intermediate forms.
Since we can’t definitively describe how eukaryotic life progressed from ancestral forms, even deducing hierarchy solely within the eukaryotic tree is difficult. In fact, the eukaryotic tree of life is in such a constant state of flux that it might as well be called a topiary, with competing sets of hands reaching for the shears.
"There’s this huge debate going on about how to root the eukaryotic tree," says Fritz-Laylin. In the absence of definitive data, people may sometimes rely on gut instinct and personal preference. "What you would want is something that allows you to discriminate between one root or another," says Cande. Naegleria itself, unfortunately, does not wield that kind of power, because more organisms are needed to get a sense of evolutionary context. "If we had more organisms to look at, and more genomes," Cande adds, "we can have a better sense of it."
More searchlights needed
As it turns out, recordkeeping is not one of Nature’s prerogatives; she’s just an accidental historian. The DNA transcript has provided many insights into life’s evolutionary history, but that information is not always easy to get. Major problems, like the abundance of lateral gene transfer in prokaryotes and some eukaryotes, make reconstructing the tree of life seem impossibly difficult. One can’t help but pose the question, "will we ever know?"
For sparsely sampled parts of the tree, like the sections Cande is investigating, the most crucial step might simply be more sequencing. Certainly the cost of genome sequencing is dropping rapidly. Two years ago, the Monosiga sequence cost approximately $300k, but the upcoming Salpingoeca genome will only set the King lab back about $40,000. The first human genome was a highly collaborative effort spanning ten years and costing over $500 million. These days you can get your genome sequenced by Illumina for a mere $15,000. That means even researchers with relatively limited funding can get their hands on a significant slice of the data pie.
The Department of Energy’s Joint Genome Institute, which is located partly at Lawrence Berkeley National Laboratory, was involved in the sequencing of both Naegleria and Monosiga and is ramping up plans to sequence more microbial genomes. In collaboration with Cande, they are also sequencing Spironucleus vortens, which is similar to Giardia in what it’s missing. Given that these organisms are only related very distantly (akin to the relationship between sea urchins and humans), the confirmation of shared characteristics will be useful in understanding whether Giardia‘s genomic minimalism is really a hallmark of early eukaryotes or simply an artifact of parasitism.
Cheaper sequencing is only part of the puzzle. Often the bigger challenge is simply collecting enough raw genetic material. Many free-living microbes are too difficult to grow in the lab. The web of life at that scale is delicate and can be impossibly complicated to replicate in vitro. Even if a species’ food source can be identified, it may be technically challenging to separate the species of interest from its prey, thus complicating genetic analyses. The proliferation of automated, high-throughput technologies may help future researchers quickly identify the proper culture conditions to grow and purify these fussy microbes.
Our technological future is poised to reveal much about our evolutionary past. However, the paramount goal is not really to catalog all of life’s genomic sequences in exquisitely arranged phylogenetic trees. Rather, both genomic sequencing and phylogenetics can be considered colossal searchlights, scanning the landscape of biological complexity for salient features to be investigated further. Together, they illuminate a much broader field.
The following are inset boxes with supplemental background information. They are also found in the print edition of the article.
The shotgun approach
 Credit: Graphics: Amy Orsborn; Data: Gregory, T. R. (2005), Animal Genome Size Database (GenomeSize.com) and Rob Carlson (Synthesis.cc)
Credit: Graphics: Amy Orsborn; Data: Gregory, T. R. (2005), Animal Genome Size Database (GenomeSize.com) and Rob Carlson (Synthesis.cc)
In 1994, Craig Venter and his colleagues applied for an NIH grant to sequence the genome of Haemophilus influenzae using a then-untested technique called "whole genome shotgun sequencing." At that time, the NIH was already four years into the Human Genome Project using a different methodology that systematically divides the genome into manageable pieces to be sequenced individually. The team of experts reviewing Venter’s application deemed it unfeasible, believing the technique would produce a hopeless jumble of data that would be impossible to piece together.
Little did the NIH know that the gun was already loaded. Only a month after the NIH rejection letter arrived, Science published Venter’s complete sequence of the H. influenzae genome, the first complete genome ever to be deciphered. Since then, genome sequencing has experienced explosive growth, largely due to the rapid efficiency of the whole genome shotgun method.
Exactly what does shotgun sequencing entail? The difference lies mostly in the logistics, and not the physics, of the sequencing. Both the NIH and Venter were using variants of Sanger chain-termination sequencing. In this method, short DNA segments are elongated using DNA polymerase in the presence of normal deoxynucleotides, the individual subunits that make up a long strand, and fluorescently labeled dideoxyribonucleotides, which are special versions of deoxyribonucleotides. Random incorporation of the dideoxynucleotide caps the molecule and prevents further elongation. Thus, the reaction makes many partial copies of the original sequence, with each copy terminated in a fluorescently labeled nucleotide. All the copies are then size-separated with very good resolution such that chains with a single base pair difference are separated. Since the four types of dideoxyribonucleotides - A, T, C, and G - are tagged with different colors, scientists can decipher the sequence simply by reading off the colors.
The limit is that Sanger sequencing only works for short strands under several hundred base pairs (bp). However, H. influenzae‘s genome is almost two million bp long and the human genome is over six billion bp long. The NIH tackled this problem by adopting the most straightforward approach - to neatly subdivide the giant tangle of genomic DNA into small parcels for sequential sequencing. However, the necessity of painstaking a priori annotation made this process excruciatingly slow. By contrast, the solution adopted by Venter’s team required very little pre-processing. They simply generated many random, but similarly sized, fragments of the genome and sequenced all of them. Then, using computational methods that take into account the size of the fragment and other technical features, they could align all the fragments and back-compute the entire sequence. Even the sequence of the human genome, which is 3,000 times the size of H. influenzae‘s genome, was eventually completed using this technique.
Venter’s experimental validation of shotgun sequencing is only part of the story. The success of whole genome sequencing is reliant upon a vast suite of diverse technologies, ranging from biochemical tags to automation to computational processing algorithms. Though shotgun sequencing has brought us a long way, so-called "next-generation" sequencing techniques promise even faster and cheaper results by sequencing arrays of many DNA strands in parallel.
This article is part of the Spring 2010 issue.